All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a healthcare professional. If you are a patient or carer, please visit the MDS Alliance.
The MDS Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the MDS Hub cannot guarantee the accuracy of translated content. The MDS Hub and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.


Now you can support HCPs in making informed decisions for their patients
Your contribution helps us continuously deliver expertly curated content to HCPs worldwide. You will also have the opportunity to make a content suggestion for consideration and receive updates on the impact contributions are making to our content.
Find out more
Create an account to access:
Bookmark & personalize site content
Receive alerts for new content in your areas of interest
View MDS content recommended for you
The challenge of diagnosing low-grade myelodysplastic syndromes
Myelodysplastic syndromes (MDS) are a heterogeneous group of myeloid neoplasms. MDS are marked by morphologic dysplasia of the different cell lines and inefficient hematopoiesis, resulting in peripheral blood cytopenias. They are characterized by heterogeneous clinical behavior, and a large number of them have the potential of transformation into acute myeloid leukemia (AML). Low-grade MDS cases lacking excess blasts present a difficult diagnostic challenge requiring a multifactorial approach, involving clinical information, laboratory and biopsy findings, as well as flow cytometry and molecular studies as needed.
In a review article recently published in the American Journal of Clinical Pathology, Siddon and Hasserjian discuss the diagnostic approach for low-grade MDS in patients lacking increased blasts, including a diagnostic algorithm and two case studies.1
A diagnostic algorithm for low-grade MDS
Due to the myriad of nonneoplastic causes of cytopenia, the differential diagnosis of MDS often requires interval re-biopsy to enable final confirmation and subsequent treatment planning. MDS evaluation requires integration of peripheral blood findings, bone marrow assessment, flow cytometry, cytogenetics, and molecular genetic studies, which can be difficult to consolidate. A diagnostic algorithm to assist with the classification of low-grade MDS on the basis of morphology and genetic testing results, based on the 2015 World Health Organization (WHO) classification of myelodysplastic syndromes, can be observed in Figure 1.
Figure 1. Diagnostic algorithm for the classification of low-grade myelodysplastic syndromes (lacking excess blasts)1
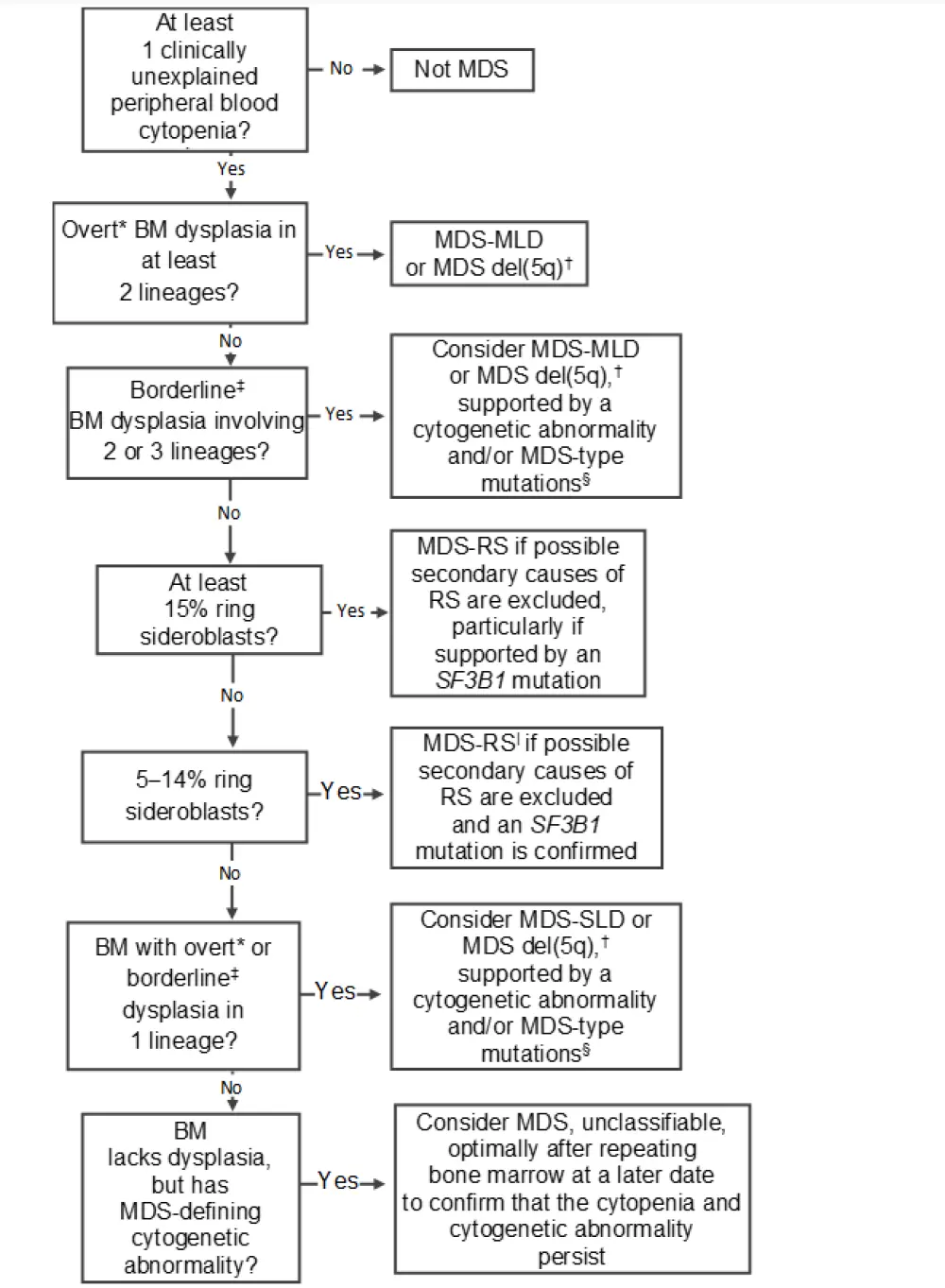
BM, bone marrow; MDS, myelodysplastic syndromes; MDS del(5q), MDS with isolated del(5q); MDS-MLD, MDS with multilineage dysplasia; MDS-RS, MDS with ring sideroblasts; MDS-SLD, MDS with single-lineage dysplasia; RS, ring sideroblasts.
*Overt dysplasia in at least 20% of erythroids/granulocytes and at least 30% of megakaryocytes displaying dysplastic changes; any true micromegakaryocytes (size of a promyelocyte or less) could constitute overt dysplasia.
†At least two incidences of 1% peripheral blood blasts could indicate a diagnosis of MDS unclassifiable. An isolated del(5q) cytogenetic abnormality and fulfillment of other features could lead to a diagnosis of MDS with isolated del(5q).
‡Borderline dysplasia = dysplasia close to 10% of the lineage.
§Caution should be exercised with: a normal karyotype and nil identified mutations; or only a single clonal hematopoiesis of indeterminate potential– type mutation at low variant allele fraction is identified; a descriptive diagnosis with a recommendation to repeat the BM examination may be indicated.
|Pancytopenia and single-lineage dysplasia = diagnosis of MDS, unclassifiable.
Due to the associated cytopenias, patients with MDS may have varied symptomatology. In order to diagnose MDS, at least one cytopenia is mandatory, however it is common to see more than one. As shown in Table 1, the most common cytopenia is anemia, reported in up to 85% of MDS cases, and the least common is pancytopenia, occurring in only 15% of patients. Subsequently, the most common patient-reported symptoms of MDS are fatigue, pallor, and/or weakness. The International Prognostic Scoring System (IPSS) gives prognostic benchmarks for cytopenias, however in MDS, diagnosis can be made with mild cytopenia in patients with indicative morphologic or cytogenetic characteristics.
Table 1. Peripheral blood findings in patients with low-grade MDS1
|
Blood Finding |
Frequency, |
Blood results |
Morphology |
|---|---|---|---|
|
Anemia |
85 |
Hemoglobin less than institutional reference range (e.g., < 10 g/dL) |
Macrocytosis |
|
Neutropenia |
40 |
Absolute neutrophil count < 1.8 × 109 /L |
Nuclear hyposegmentation |
|
Thrombocytopenia |
30–40 |
Platelets less than institutional reference range |
Platelet hypogranularity |
|
Pancytopenia |
15 |
All of the above |
|
The evaluation of patients for possible MDS usually begins with examination of a peripheral blood smear. In low-grade MDS, this varies from isolated anemia (usually macrocytic) with unremarkable red blood cell morphology, through to remarkable red blood cell anisopoikilocytosis and basophilic stippling. Abnormal leukocyte morphology may be present appearing as hyposegmented neutrophils (pseudo Pelger–Huët cells), that may or may not possess cytoplasmic hypogranularity.
In general, a core biopsy histology of low-grade MDS reveals hypercellularity relative to the age of the patient. Hypocellular marrows can be found in a small number of patients with MDS, more frequently in pediatric patients and in MDS following cytotoxic therapy. In MDS, there may be aberrant localization of immature precursor cells, where clusters of blasts are found away from bone trabeculae. The lineage of the dysplasia is assessed primarily on the bone marrow aspirate, in the case of erythroid or myeloid disease, while megakaryocyte dysplasia can be detected by either the core biopsy specimen or aspirate. In granulocytic dysplasia, the aspirate most commonly exhibits nuclear hypolobation and/or a subset of neutrophils with cytoplasmic hypogranularity.
At least 10% of a lineage is required to be dysplastic for a diagnosis of MDS. This must be viewed in the context of other causes of secondary morphologic dysplasia, such as medications, infections, nutritional deficiencies, toxins, bone marrow lymphoma, plasma cell neoplasms, and autoimmune diseases (Table 2). The remarkable interobserver variability in diagnosing patients with MDS reaches its peak among patients with low-grade MDS compared with MDS with excess blasts. This could be due to the fact that, in some low-grade MDS cases, a few or no overt morphologic dysplasia may be reported. Additional stains such as CD61 (for megakaryocytes), reticulin (increased fibrosis), and CD34 (CD34+ blasts) may assist in both diagnosis and prognosis.
Table 2. Causes of secondary dysplasia and the associated morphologic features
|
EBV, Epstein-Barr virus; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; HIV, human immunodeficiency virus. |
||
|
Secondary Cause |
Specific Causes |
Morphologic Findings |
|---|---|---|
|
Nutritional deficiency |
Vitamin B12/folate, copper |
Megaloblastic anemia, giant bands and metamyelocytes, myeloid hypersegmentation, erythroid vacuolization |
|
Autoimmune disorders |
Rheumatoid arthritis, lupus erythematosus |
Hypocellularity, fibrosis |
|
Viral infections |
HIV, hepatitis B/C, EBV |
Hypocellularity, myeloid left shift, lymphoid aggregates, dysplasia in erythroid and megakaryocytic cells |
|
Cytokines |
G-CSF, GM-CSF |
Myeloid left shift, toxic granulation, increased blasts |
|
Medications |
Tacrolimus, valproic acid, ganciclovir, mycophenolate mofetil, azathioprine, isoniazid, chloramphenicol, trimethoprim |
Hypocellularity, dyserythropoiesis, ring sideroblasts, mild reticulin fibrosis, dysgranulopoiesis (specifically with valproic acid) |
|
Toxins |
Alcohol, benzene, arsenic |
Dyserythropoiesis, ring sideroblasts |
|
Paroxysmal nocturnal hemoglobinuria |
Acquired |
Erythroid predominance, dyserythropoiesis |
|
Erythroid stress response |
Acute blood loss, hemoglobinopathies, autoimmune hemolytic anemia |
Erythroid hyperplasia, dyserythropoiesis |
Flow cytometric assays can assist the diagnosis of patients with MDS, especially in those subtle low-grade cases. Flow cytometry can not only give blast percentage, but also important information regarding the overall immunophenotype of the blasts, like aberrant expression of CD5, CD7, CD19, and/or CD56. The European LeukemiaNet Working Group suggest that in patients without significant morphologic or cytogenetic findings indicative of MDS, three or four flow cytometric abnormalities should trigger repeat bone marrow evaluation under suspicion of evolving MDS. However, a lack of determinative immunophenotype between MDS and non-MDS conditions has limited the application of flow cytometry in this disease.
Historically, conventional karyotyping has been used for both diagnosis and prognosis of MDS using the IPSS. However, chromosome abnormalities are seen in only 50% of new MDS diagnoses, being less common in low-risk MDS subtypes, such as MDS with ring sideroblasts and MDS with single-lineage dysplasia. Clonality of specific karyotype abnormalities can assist MDS diagnosis even in the absence of definitive morphologic dysplasia. Unfortunately, a number of the most common cytogenetic abnormalities reported in MDS, –Y, +8, and del(20q), have also been reported in nonneoplastic conditions, e.g., aplastic anemia, and are not considered differential in the diagnosis of MDS without adequate morphologic dysplasia. Fluorescence in situ hybridization can assist in confirming karyotypic abnormalities but is not necessary if conventional methods have been used in at least 20 metaphase cells. MDS with isolated del(5q) is a distinct WHO category based on a karyotype abnormality. This is characterized by anemia and thrombocytosis with bone marrow characteristics, including many small, hypolobated megakaryocytes and no increase in blasts.
Molecular techniques, such as high-throughput sequencing technologies, can assist diagnostic profiling by providing novel information about acquired DNA variants in myeloid neoplasms. Although high-throughput sequencing technologies are not currently applied for diagnostic purposes alone, DNA variants could assist in understanding pathogenesis, prognosis, and targeted treatment options in MDS.
Conclusion
The diagnosis of low-grade MDS represents one of the most challenging hematopathology topics due to high interobserver variability in the assessment of dysplasia morphology and a myriad of non-MDS causes of cytopenia and dysplasia in elderly patients. Additionally, MDS evaluation requires integrative data, which incorporates peripheral blood findings, assessment of bone marrow, flow cytometry, cytogenetic studies, and possibly molecular genetic studies. These data can be difficult to consolidate, but as the cases below demonstrate, such endeavor is necessary to delineate a comprehensive morphologic, clinical, and genetic diagnosis in, for example, the complex clinical context of worsening and unexplained anemia.
Case Study 1
Presentation
A 68-year-old man with long-standing untreated isolated anemia (hemoglobin [Hb], 8.1 g/dL [reference range (RR), 12.0–18.0]; mean corpuscular volume [MCV], 95.1 fL [RR, 78.0–94.0]). The anemia had recently worsened and had been treated with epoetin alfa.
Diagnosis
Due to the significant chronic anemia, a diagnosis of MDS was taken into consideration. Analysis of bone marrow aspirate revealed a hypercellular, erythroid predominant marrow with dyserythropoiesis, including megaloblastoid changes, nuclear budding, and irregular nuclear contours without ring sideroblasts on an iron stain. No remarkable morphological changes were seen in the myeloid and megakaryocyte lineages. There was no obvious increase in blast count either morphologically or by flow cytometry. Cytogenetic analysis found a normal male karyotype in 20 metaphase cells. A single pathogenic DNMT3A variant found at 4% variant allelic frequency was reported in next-generation sequencing (NGS) panel.
Outcome
Not MDS
Further history reveals hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease) with frequent epistaxis causing anemia. Erythroid predominance and dysplasia in the marrow are secondary changes due to the frequent and significant bleeding episodes. The single DNMT3A variant is likely an incidental mutation in the absence of evidence of primary myelodysplasia or other clonal abnormalities.
Case Study 2
Presentation
An 84-year-old man had symptomatic isolated anemia with normal iron studies (Hb, 7.3 g/dL [RR, 13.5–17.5]; MCV, 76.9 fL [RR, 80.0–100.0]). He presented with a history of anemia but new onset of fatigue.
Diagnosis
Analysis of bone marrow aspirate revealed a hypercellular marrow for his age with erythroid predominance, left shift, and notable dyserythropoiesis manifesting as nuclear irregularities. There was no dysplasia in the granulocytes or megakaryocytes. Iron staining showed ring sideroblasts comprising 25% of the erythroid fraction. Hemoglobin electrophoresis revealed increased hemoglobin A2 at 5.4% (RR, 2.0–3.3%) and hemoglobin F at 2.7% (RR, 0–0.9%), consistent with a diagnosis of β-thalassemia. Cytogenetic analysis reported an abnormal 46, XY, del(20)(q11.2q13.3) karyotype in 18 of 20 metaphase cells. A single pathogenic SF3B1 variant present at 8% allelic frequency was reported in NGS.
Outcome
MDS with ring sideroblasts and single-lineage dysplasia
The known inherited cause of anemia (β-thalassemia) and microcytosis may discourage an MDS diagnosis. The examination of blood parameters indicated a recent worsening of anemia coinciding with rising MCV, making the anemia macrocytic relative to baseline MCV. While the del(20q) cytogenetic abnormality can be seen in non-MDS conditions, the SF3B1 mutation indicates a neoplastic origin for the ring sideroblasts.
References
Please indicate your level of agreement with the following statements:
The content was clear and easy to understand
The content addressed the learning objectives
The content was relevant to my practice
I will change my clinical practice as a result of this content