All content on this site is intended for healthcare professionals only. By acknowledging this message and accessing the information on this website you are confirming that you are a healthcare professional. If you are a patient or carer, please visit the MDS Alliance.
The MDS Hub website uses a third-party service provided by Google that dynamically translates web content. Translations are machine generated, so may not be an exact or complete translation, and the MDS Hub cannot guarantee the accuracy of translated content. The MDS Hub and its employees will not be liable for any direct, indirect, or consequential damages (even if foreseeable) resulting from use of the Google Translate feature. For further support with Google Translate, visit Google Translate Help.


Now you can support HCPs in making informed decisions for their patients
Your contribution helps us continuously deliver expertly curated content to HCPs worldwide. You will also have the opportunity to make a content suggestion for consideration and receive updates on the impact contributions are making to our content.
Find out more
Create an account to access:
Bookmark & personalize site content
Receive alerts for new content in your areas of interest
View MDS content recommended for you
Inherited hematologic malignancies predisposition syndromes
Hereditary predispositions to hematologic malignancies occur in approximately 5─10% of cases. During the Society of Hematologic Oncology (SOHO) Eighth Annual Meeting, Courtney DiNardo gave a talk about the importance of recognizing patients with inherited cancer predisposition syndromes.1
The identification of an inherited predisposition allows optimized treatment of the patient, more informed donor decisions (avoiding a related donor with the same predisposition), and opportunities for identification and screening of family members.1
Inherited predisposition syndromes associated with leukemia and myeloproliferative neoplasms (MPN) are reported in Table 1.
Table 1. Leukemia and MPN predisposition syndromes1
|
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; B-ALL, B-cell ALL; CLL, chronic lymphocytic leukemia; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasms; T-ALL, T-cell ALL. |
|
|
Hematologic malignancy |
Predisposition syndrome |
|
Adult bone marrow failure/ hypocellular MDS/AML |
Fanconi anemia |
|
Dyskeratosis congenita |
|
|
Shwachman–Diamond syndrome |
|
|
Diamond Blackfan anemia |
|
|
Familial aplastic anemia and hypocellular MDS |
|
|
Familial MDS/acute leukemia |
RUNX1 familial platelet disorder/AML (and T-ALL) |
|
GATA2 familial MDS/AML (MonoMAC syndrome) |
|
|
CEBPα familial AML |
|
|
ANKRD26 familial platelet disorder with propensity to MDS/AML |
|
|
DDX41 familial MDS/AML |
|
|
ETV6 familial MDS/AML/lymphoid malignancies |
|
|
SAMD9/SAMD9L familial MDS & immunodeficiency syndrome |
|
|
Familial ALL |
Li–Fraumeni syndrome: germline TP53 mutations |
|
PAX5: familial B-cell lymphoid leukemias/lymphomas |
|
|
IKZF1-associated predisposition to B-ALL |
|
|
Biallelic SH2B3 mutations (also MPN risk?) |
|
|
Familial CLL |
POT1 familial CLL/melanoma/cardiac myxomas |
|
Familial MPN |
JAK2 V617I, R564Q & R867Q/N, others |
|
|
MPL S505N, W515R, P106L, others |
|
|
TET2 D1858fs |
|
|
700 kb duplication, with ATG2B and GSKIP overexpression, leads to increased progenitor sensitivity to thrombopoietin |
In 15─20% of cases, patients with either aplastic anemia, hypocellular myelodysplastic syndrome (MDS), or bone marrow failure have an inherited predisposition syndrome, such as Fanconi anemia, dyskeratosis congenita (DC), Shwachman–Diamond syndrome, Diamond Blackfan anemia, or others. These patients should be recognized and referred for genetic testing in order to provide optimal treatment strategies.1
Genetic profiling of patients with inherited bone marrow failure syndromes is important because they
- respond poorly to anti-thymocyte globulin and cyclosporine (a shorter telomere length by flow-FISH [fluorescent in situ hybridization] in patients with aplastic anemia correlates with poor response to standard immune suppressive therapy);
- respond well to growth factors and androgens (effective in ˃ 50% of patients);
- should be treated with specialized reduced-intensity regimens to avoid excessive morbidity (avoid alkylating agents); and
- need surveillance for solid tumors.
Clinical evaluation1
Dyskeratosis congenita
DC (also called short telomere syndrome, telomere biology disorder, or telomeropathy) is caused by mutations in genes important for telomere maintenance. Telomeres protect chromosome ends from degradation, and their length regulates the ability of a cell to duplicate. The instability resulting from an inadequate telomere maintenance can lead to uncontrolled cell growth and development of cancer.
Telomere flow-FISH is the technique used to detect DC. With telomere flow-FISH it is possible to distinguish DC from other causes of bone marrow failure.
Familial GATA2 (MonoMAC) syndrome
Familial GATA2 syndrome, also known as MonoMAC or Emberger syndrome, is due to autosomal dominant mutations or deletion of GATA2 and is associated with immunodeficiency and frequent infections. The median age at diagnosis is 18 years, and the lifetime risk of MDS/acute myeloid leukemia (AML) is 75–90%. It is particularly common in younger patients with MDS, with 10─15% of them having germline GATA2 mutations, and occurs frequently in those with MDS carrying monosomy of chromosome 7:
- 27% of children and young adults
- 72% of patients aged 12─19
In cases of transplants from a related donor, it is important to test family members to be sure that they are not GATA2 mutation-positive. However, overall survival and stem cell transplant outcomes appear to be independent of GATA2 status.
RUNX1, ANKRD26, and ETV6 variants
MDS and acute leukemia predisposition syndromes include autosomal dominant pathogenic variants in RUNX1 (21q), ANKRD26 (10p12), and ETV6 (12p13), which are associated with mild to moderate thrombocytopenia, platelet aggregation defects, and abnormal megakaryocytes.
RUNX1 was the first described, and about 40% of patients with germline mutations develop MDS/AML/T-cell acute lymphoblastic leukemia at a median age of 33 years. Thus, monitoring patients found to harbor these mutations is important to identify those at risk.
DDX41 mutations
Autosomal dominant mutations in DDX41 (5q35) have been recently identified to confer a predisposition to developing MDS/AML at a median age of 65 years. DDX41 is mutated in 1% of all patients with MDS/AML, and retrospective studies have shown that these patients are particularly sensitive to lenalidomide. The majority of DDX41 mutations leading to MDS/AML are germline mutations, and therefore inherited. The estimated risk of developing MDS/AML in family members of an individual with a DDX41 mutation is ̴20─30%.
Genetic characterization of disorders
An 81-gene panel, called EndLeukemia panel, was developed in 2017 to allow comprehensive molecular profiling of a variety of hematologic malignancies.2 Genes included in the panel, reported in Table 2, cover genetic alterations in AML, MDS, MPN, MDS/MPN, aplastic anemia, acute lymphoblastic leukemia, hairy cell leukemia, T-large granular lymphocytic leukemia, and T-prolymphocytic leukemia.2
When suspected germline variants are detected, a multidisciplinary review is required to identify patients that should be further evaluated; the workflow is reported in Figure 1.1,3
Table 2. Genes included in the EndLeukemia panel1
|
Chromosome |
Gene |
|
1 |
BRINP3, CSF3R, GFI1, JAK1, MPL, NRAS, |
|
2 |
ASXL2, DNMT3A, IDH1, SF3B1 |
|
3 |
CBLB, GATA2, STAG1, TERC |
|
4 |
FBXW7, KIT, TET2 |
|
5 |
DDX41, IL7R, NPM1, TERT |
|
7 |
BRAF, CUX1, EZH2, IKZF1 |
|
8 |
RAD21 |
|
9 |
HNRNPK, JAK2, NOTCH1, PAX5 |
|
10 |
ANKRD26, PTEN, SMC3 |
|
11 |
CBL, EED, HRAS, MLL, SF1, WT1 |
|
12 |
ETNK1, ETV6, KRAS, PTPN11, PRPF40B, SH2B3 |
|
13 |
FLT3 |
|
15 |
IDH2, MAP2K1, PML |
|
16 |
CREBBP |
|
17 |
NF1, RARA, SRSF2, STAT3, STAT5A, STAT5B, SUZ12, TP53 |
|
18 |
SETBP1 |
|
19 |
CALR, CBLC, CEBPA, ELANE, JAK3, U2AF2 |
|
20 |
ASXL1, GNAS |
|
21 |
RUNX1, U2AF1 |
|
22 |
SF3A1 |
|
X |
BCOR, BCORL1, CRLF2, GATA1, IL2RG, KDM6A, PHF6, PIGA, SMC1A, STAG2, ZRSR2 |
Figure 1. Workflow for the multidisciplinary review of suspected germline variants3
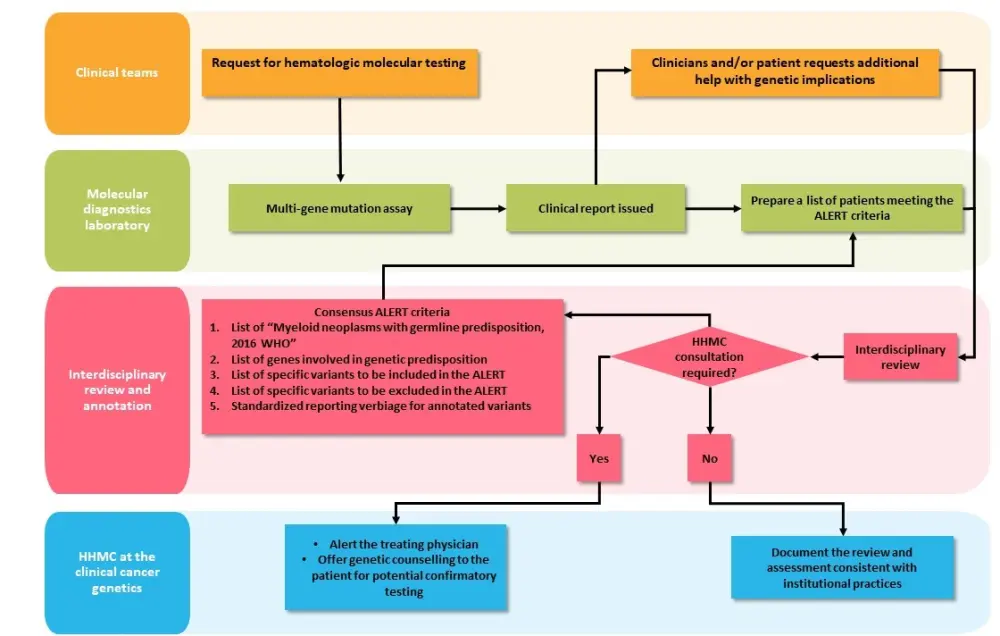
HHMC, hereditary hematologic malignancies clinic; WHO, World Health Organization.
Current consensus statement1
The recommendations for clinicians managing patients with familial hematologic malignancies syndrome are the following:
- Clinical exam with complete blood count one or two times a year;
- Bone marrow biopsy at the time of diagnosis, to identify any clonal changes or secondary mutations that may lead to the development of hematologic malignancies, and then every year for research surveillance;
- Consider allogeneic stem cell transplant at time of development of dysplasia, or clonal somatic cytogenetic or molecular abnormality; and
- For younger and interested patients, suggest reproductive counseling and preimplantation genetic testing.
References
Please indicate your level of agreement with the following statements:
The content was clear and easy to understand
The content addressed the learning objectives
The content was relevant to my practice
I will change my clinical practice as a result of this content